Volume 4, Issue 10
October 2024
Obesity-Induced Inflammation and Its Role in the Development of Insulin Resistance
Marwah Yakoop Abdullah, Saad Daham AlQwaidi, Aisha Mohammed Alshehri, Fatimah Ali Alamry, Raghad Saad Alamri, Zainab Khaleel Alsolbi, Bushra Haidar Alqurashi, Yousef Ali Aldandan, Awadh Muhaysin Alrashidi, Maha Ayed Aljumaie
DOI: http://dx.doi.org/10.52533/JOHS.2024.41002
Keywords: obesity, inflammation, insulin resistance, adipose tissue dysfunction, pro-inflammatory cytokines
Obesity is a leading global health issue closely associated with metabolic disorders, including insulin resistance and Type 2 Diabetes Mellitus (T2DM). A central mechanism linking obesity to these metabolic dysfunctions is chronic, low-grade inflammation. In individuals with obesity, adipose tissue becomes dysfunctional, characterized by adipocyte hypertrophy, hypoxia, and the infiltration of immune cells such as macrophages. These changes lead to the release of pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and monocyte chemoattractant protein-1 (MCP-1), which interfere with insulin signaling pathways. The activation of inflammatory pathways like nuclear factor-kappa B (NF-κB) and c-Jun N-terminal kinase (JNK) promotes the serine phosphorylation of insulin receptor substrates (IRS), impairing their ability to propagate insulin signals effectively. This disruption leads to reduced glucose uptake in tissues such as the liver, muscle, and adipose tissue, contributing to systemic insulin resistance. Additionally, the NLRP3 inflammasome pathway exacerbates inflammation by activating caspase-1 and increasing IL-1β production, further impairing insulin signaling. Free fatty acids (FFAs) released from hypertrophic adipocytes also play a significant role in perpetuating inflammation and disrupting insulin sensitivity. FFAs activate toll-like receptor 4 (TLR4) on immune cells, enhancing the inflammatory response and contributing to ectopic fat deposition in non-adipose tissues, further promoting insulin resistance. Understanding the mechanisms linking obesity-induced inflammation to insulin resistance provides insight into potential therapeutic interventions. Targeting chronic inflammation, modulating cytokine production, and addressing adipose tissue dysfunction may improve insulin sensitivity and help manage obesity-related metabolic disorders. This highlights the need for a multifaceted approach to treat obesity and its associated complications, particularly focusing on reducing inflammation to mitigate the risk of insulin resistance and T2DM.
Introduction
Obesity has become a significant global health concern, linked to various metabolic disorders, including insulin resistance, Type 2 Diabetes Mellitus (T2DM), cardiovascular diseases, and non-alcoholic fatty liver disease (1). The prevalence of obesity has increased dramatically in recent decades, mirroring a rise in these metabolic diseases. Among the most pressing complications associated with obesity is insulin resistance, a condition in which the body’s cells, particularly in muscle, liver, and fat tissue, fail to respond adequately to insulin. This impaired insulin signaling disrupts glucose homeostasis, leading to elevated blood sugar levels and compensatory insulin production. Over time, this cycle can culminate in T2DM (2).
Inflammation has been identified as a central mechanism connecting obesity to insulin resistance. Obesity-induced inflammation refers to a chronic, low-grade inflammatory state that arises as adipose tissue accumulates beyond its functional capacity. Adipose tissue, traditionally seen as a mere fat storage site, also plays an active role in metabolism by secreting bioactive substances called adipokines. These include hormones and cytokines that regulate glucose metabolism, lipid homeostasis, and immune responses. However, in obesity, there is a shift in adipose tissue function, resulting in the secretion of pro-inflammatory adipokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and monocyte chemoattractant protein-1 (MCP-1) (3). This shift drives a state of chronic inflammation that disrupts normal insulin signaling pathways and contributes to the development of insulin resistance.
Key inflammatory pathways, such as the c-Jun N-terminal kinase (JNK) and nuclear factor-kappa B (NF-κB) pathways, are particularly important in the pathogenesis of insulin resistance. These pathways are activated by inflammatory cytokines and stress signals, leading to the phosphorylation of insulin receptor substrates (IRS). This phosphorylation impairs the IRS's ability to propagate insulin signals, causing reduced glucose uptake in muscle and fat cells and increased glucose production in the liver, which worsens hyperglycemia and insulin resistance (4). Moreover, obesity triggers the recruitment of immune cells, particularly macrophages, into adipose tissue. This process amplifies the inflammatory response. In lean individuals, adipose tissue primarily contains anti-inflammatory M2 macrophages, whereas in obesity, there is a shift toward pro-inflammatory M1 macrophages. These M1 macrophages secrete cytokines that further disrupt insulin signaling and exacerbate metabolic dysfunction (5). Another significant factor linking obesity to inflammation and insulin resistance is adipose tissue dysfunction. As adipocytes (fat cells) expand in response to excess nutrient intake, they undergo structural and functional changes, including hypertrophy, hypoxia, and endoplasmic reticulum stress. Hypertrophic adipocytes lose their capacity for effective insulin signaling, leading to the release of higher levels of free fatty acids (FFAs). Elevated circulating FFAs have been shown to contribute to insulin resistance by promoting the activation of toll-like receptor 4 (TLR4) on immune cells, which triggers pro-inflammatory pathways. This creates a vicious cycle where adipose tissue dysfunction fuels inflammation, which in turn impairs insulin action (3).
Hypoxia, a condition in which expanding adipose tissue does not receive adequate oxygen, is another contributor to obesity-induced inflammation. Hypoxia results in necrotic cell death within adipose tissue, triggering an immune response that promotes the infiltration of macrophages and the release of pro-inflammatory mediators. This further impairs insulin signaling, contributing to the progression of insulin resistance (2). Additionally, endoplasmic reticulum stress in obese adipose tissue plays a role in triggering inflammation. The endoplasmic reticulum is responsible for proper protein folding, but in obesity, excessive nutrient intake overwhelms this process, leading to the accumulation of misfolded proteins. This activates the unfolded protein response (UPR), a cellular stress response that promotes inflammation and contributes to metabolic dysregulation. These intertwined mechanisms highlight the critical role of obesity-induced inflammation in the development of insulin resistance. Chronic inflammation in adipose tissue, driven by an imbalance of pro-inflammatory adipokines, immune cell infiltration, and adipocyte dysfunction, plays a key role in disrupting insulin signaling. Understanding these pathways provides valuable insight into the relationship between obesity and metabolic disorders and informs potential therapeutic interventions aimed at reducing inflammation to improve insulin sensitivity and prevent the progression of related diseases.
Review
Obesity-induced inflammation is a multifaceted process that plays a pivotal role in the development of insulin resistance. As adipose tissue expands, its secretory function becomes dysregulated, leading to an overproduction of pro-inflammatory cytokines, such as TNF-α and IL-6. These cytokines not only impair insulin signaling but also activate stress-related pathways, such as the JNK and NF-κB pathways, which further contribute to metabolic dysfunction (6). This low-grade, chronic inflammatory state disrupts glucose homeostasis by interfering with the IRS and inhibiting glucose uptake in tissues like the liver, muscle, and fat. Moreover, immune cells, particularly macrophages, are recruited to adipose tissue in response to the inflammatory environment. This shift from anti-inflammatory (M2) to pro-inflammatory (M1) macrophages exacerbates insulin resistance. The inflammatory state is further aggravated by adipocyte hypertrophy and hypoxia, which contribute to increased release of free fatty acids (7). Free fatty acids, in turn, activate TLR4, enhancing inflammation and creating a vicious cycle that perpetuates insulin resistance. These interconnected mechanisms underscore the importance of addressing inflammation as a therapeutic target for improving insulin sensitivity and managing obesity-related metabolic disorders.
Mechanisms Linking Obesity and Chronic Inflammation
The connection between obesity and chronic inflammation is complex, with multiple molecular and cellular mechanisms contributing to this relationship. One of the primary drivers of inflammation in obesity is the dysfunction of adipose tissue. As adipocytes expand due to excess nutrient intake, they become hypertrophic and experience stress, which leads to the release of pro-inflammatory molecules such as IL-6 and TNF-α (7, 8). This inflammatory response originates within the adipose tissue but can spread systemically, affecting other organs and tissues involved in glucose metabolism, such as the liver and skeletal muscles.
The infiltration of immune cells, particularly macrophages, into adipose tissue plays a crucial role in chronic inflammation. In lean individuals, adipose tissue contains a higher proportion of M2 macrophages, which have anti-inflammatory properties. However, in obesity, there is a shift toward M1 macrophages, which produce pro-inflammatory cytokines that amplify the inflammatory state (9). This macrophage infiltration is often triggered by dying or necrotic adipocytes in obese tissue, which release danger signals that attract immune cells. The increased presence of M1 macrophages exacerbates local inflammation, leading to further insulin resistance and metabolic disturbances. Another mechanism linking obesity to inflammation is the role of FFAs.
In obese individuals, there is an excessive release of FFAs from adipose tissue into the bloodstream. Elevated FFAs can directly activate inflammatory pathways by binding to toll-like receptors (TLRs) on macrophages and other immune cells. This activation leads to the production of pro-inflammatory cytokines, such as IL-1β, and enhances the inflammatory response (10). FFAs promote insulin resistance by interfering with insulin signaling in tissues like the liver and muscle, contributing to systemic metabolic dysfunction. Endoplasmic reticulum stress is another critical factor that links obesity to chronic inflammation. As adipocytes expand, the demand for protein synthesis increases, leading to endoplasmic reticulum stress. The unfolded protein response (UPR) is activated in response to this stress, and while initially protective, prolonged UPR activation can promote inflammation by triggering inflammatory signaling pathways such as the NF-κB pathway. This further exacerbates the inflammatory state in obese individuals, contributing to the development of insulin resistance and metabolic syndrome (11).
Inflammatory Pathways Contributing to Insulin Resistance
Several inflammatory pathways are activated in obesity, contributing significantly to the development of insulin resistance. One of the most well-studied pathways is the NF-κB pathway. NF-κB is a transcription factor that plays a key role in regulating the expression of pro-inflammatory cytokines such as TNF-α, IL-6, and interleukin-1β (IL-1β). In obesity, these cytokines activate NF-κB through receptors such as TLR4, which recognizes FFAs released from adipose tissue (12). The activation of NF-κB leads to a chronic inflammatory response that interferes with insulin signaling by promoting the phosphorylation of IRS, reducing their ability to transmit insulin signals to downstream pathways, such as the phosphoinositide 3-kinase (PI3K)-Akt pathway, crucial for glucose uptake.
Another important inflammatory pathway implicated in insulin resistance is the JNK pathway. JNK is a stress-activated protein kinase that responds to various cellular stressors, including inflammation, endoplasmic reticulum stress, and nutrient overload, all common in obesity. The activation of JNK leads to the phosphorylation of serine residues on IRS proteins, which impairs insulin receptor signaling (13). This reduces insulin-stimulated glucose transport into cells, thereby contributing to hyperglycemia and insulin resistance. In addition to directly impairing insulin signaling, JNK also promotes the expression of pro-inflammatory cytokines, further amplifying the inflammatory response in adipose tissue and other insulin-sensitive tissues.
The NLRP3 inflammasome pathway also plays a significant role in obesity-induced insulin resistance. The NLRP3 inflammasome is a multi-protein complex that senses cellular damage and stress, leading to the activation of caspase-1 and the subsequent cleavage of pro-IL-1β into its active form, IL-1β (14). Elevated levels of FFAs, mitochondrial dysfunction, and endoplasmic reticulum stress in obese adipose tissue trigger the activation of the NLRP3 inflammasome. IL-1β, in turn, exacerbates local and systemic inflammation by promoting the release of additional pro-inflammatory cytokines and chemokines. This persistent activation of the NLRP3 inflammasome in obesity has been shown to disrupt insulin signaling in various tissues, contributing to the development of insulin resistance. Collectively, these pathways—NF-κB, JNK, and NLRP3 inflammasome—create a feedback loop of chronic inflammation and impaired insulin signaling in obesity. By disrupting the normal function of insulin receptor substrates and promoting pro-inflammatory cytokine production, these pathways play a central role in the metabolic dysregulation observed in obesity-associated insulin resistance.
Adipose Tissue Dysfunction in Obesity-Induced Inflammation
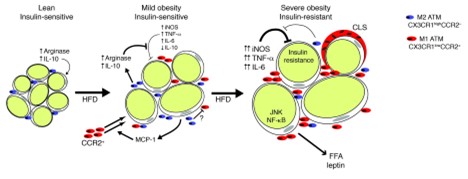
Adipose tissue dysfunction is a central contributor to inflammation in obesity, leading to metabolic complications such as insulin resistance. In lean individuals, adipose tissue serves as an efficient storage site for lipids and secretes anti-inflammatory adipokines like adiponectin, which enhances insulin sensitivity. However, as obesity develops, adipose tissue undergoes several changes, including hypertrophy and hyperplasia of adipocytes, which disrupt its function (14, 15). According to Lumeng et al., adipose tissue macrophages infiltrate adipose tissue during obesity and contribute to insulin resistance (Figure 1) (16). Hypertrophic adipocytes, characteristic of obesity, experience hypoxia due to inadequate vascularization, leading to cellular stress and necrosis. The dying adipocytes release danger signals that recruit immune cells, particularly macrophages, into the tissue, further exacerbating the inflammatory response. The recruitment of macrophages into obese adipose tissue marks a key event in the development of chronic inflammation. In healthy tissue, the resident macrophages are predominantly of the M2 phenotype, which promotes anti-inflammatory responses and tissue repair. However, in obesity, there is a shift towards the M1 macrophage phenotype, which secretes pro-inflammatory cytokines such as TNF-α, IL-6, and MCP-1 (16). These cytokines further impair insulin signaling by activating inflammatory pathways like NF-κB and JNK, both of which interfere with insulin receptor substrate (IRS) function. The result is a reduction in insulin-stimulated glucose uptake in adipose tissue, muscle, and liver, contributing to systemic insulin resistance.
Figure 1: Proposed model for adipose tissue macrophages polarization and its function in adipose tissue with progressive obesity (16).
In addition to immune cell infiltration, the secretion of FFAs from dysfunctional adipose tissue plays a significant role in promoting inflammation. Adipocytes in obese individuals release increased amounts of FFAs into circulation, which can activate TLR4 on macrophages and other immune cells. This activation triggers pro-inflammatory signaling pathways, leading to further production of cytokines like IL-1β and IL-18, both of which impair insulin signaling (17). Moreover, FFAs contribute to the development of ectopic fat deposition in non-adipose tissues, such as the liver and skeletal muscles, where they promote local inflammation and metabolic dysfunction. Another important aspect of adipose tissue dysfunction in obesity is the activation of endoplasmic reticulum stress. In hypertrophic adipocytes, the excessive accumulation of lipids and proteins overwhelms the endoplasmic reticulum capacity to fold proteins properly, leading to the activation of the UPR. While initially protective, chronic UPR activation induces inflammation through signaling pathways such as NF-κB and JNK, further disrupting insulin signaling and promoting insulin resistance.
Impact of Inflammatory Cytokines on Insulin Signaling Pathways
Inflammatory cytokines play a critical role in disrupting insulin signaling pathways, particularly in the context of obesity-induced inflammation. Among these, TNF-α is one of the most influential cytokines, with its levels markedly elevated in the adipose tissue of obese individuals. TNF-α impairs insulin signaling by promoting the serine phosphorylation of IRS, specifically IRS-1, which is a key mediator in the insulin signaling cascade. This phosphorylation reduces the ability of IRS-1 to activate downstream effectors such as PI3K and Akt, ultimately leading to decreased glucose uptake in insulin-sensitive tissues (18, 19). As a result, insulin resistance develops, with the body’s normal mechanisms for glucose metabolism becoming impaired, contributing to the hyperglycemia commonly observed in obese individuals.
IL-6 is another pro-inflammatory cytokine that negatively affects insulin signaling. While IL-6 has both anti-inflammatory and pro-inflammatory properties depending on the context, chronic elevation of IL-6 in obesity is associated with impaired insulin sensitivity. IL-6 interferes with insulin signaling by activating the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, which competes with the insulin receptor signaling pathway (20). This competition inhibits the insulin receptor's ability to effectively propagate its signal through IRS-1 and PI3K, reducing glucose transport into muscle and adipose tissue. Additionally, IL-6 induces the production of suppressor of cytokine signaling-3 (SOCS-3), which further inhibits insulin receptor signaling by promoting the degradation of IRS proteins.
MCP-1 also plays a significant role in the inflammation-mediated disruption of insulin signaling. MCP-1 is primarily responsible for the recruitment of macrophages into adipose tissue in obesity, where these immune cells contribute to the inflammatory milieu by releasing additional cytokines like TNF-α and IL-6. Elevated levels of MCP-1 correlate with increased insulin resistance, as MCP-1 indirectly enhances cytokine-mediated inhibition of the insulin signaling pathway (21). Moreover, MCP-1-induced macrophage infiltration amplifies the local inflammatory response, creating a feedback loop that perpetuates cytokine production and further impairs insulin action. The combined effect of these cytokines—TNF-α, IL-6, and MCP-1—on the insulin signaling pathway illustrates the complex interplay between chronic inflammation and metabolic dysfunction in obesity (21). By inhibiting key steps in the insulin signaling cascade, these cytokines create an environment where insulin resistance can thrive, further exacerbating the metabolic complications associated with obesity.
Conclusion
Obesity-induced inflammation plays a pivotal role in the development of insulin resistance through the activation of various inflammatory pathways, such as NF-κB, JNK, and NLRP3 inflammasome. Dysregulated adipose tissue function, macrophage infiltration, and elevated pro-inflammatory cytokines like TNF-α, IL-6 and MCP-1 contribute to impaired insulin signaling. Understanding these mechanisms is crucial for developing targeted therapies aimed at reducing inflammation and improving insulin sensitivity in obesity-related metabolic disorders. Addressing both inflammation and metabolic dysfunction may offer significant potential in managing and preventing the progression of insulin resistance and its complications.
Disclosures
Author Contributions
The author has reviewed the final version to be published and agreed to be accountable for all aspects of the work.
Ethics Statement
Not applicable
Consent for publications
Not applicable
Data Availability
All data is provided within the manuscript.
Conflict of interest
The authors declare no competing interest.