Volume 4, Issue 9
September 2024
Pediatric Neurocutaneous Syndromes: Genetic Mutations, Clinical Features, and Dermatological Manifestations
Nawal Rajeh Alyamani, Noor Mohammed Alharbi, Hadeel Ayman Alrabee, Aseel Mudhhi Alotaibi, Fatima Yaqoob Mohamed, Abdulrahman Obaid Alanazi, Fatmah Abdullah Aldosari, Reema Hassan Alghmdi, Rand Fahad Alabood, Deema Faleh Alanazi
DOI: http://dx.doi.org/10.52533/JOHS.2024.40908
Keywords: pediatric neurocutaneous syndromes, genetic mutations, neurological manifestations, dermatological features, targeted therapies
Pediatric neurocutaneous syndromes are a group of genetically inherited disorders characterized by the simultaneous involvement of the skin, nervous system, and other organ systems. These syndromes, which include Neurofibromatosis Type 1 (NF1), Tuberous Sclerosis Complex (TSC), and Sturge-Weber Syndrome (SWS), arise from specific genetic mutations that disrupt normal cellular processes, leading to a wide range of clinical manifestations. The genetic mutations associated with these syndromes, such as those in the NF1, TSC1, TSC2, and GNAQ genes, result in dysregulated cell growth and differentiation, contributing to the development of tumors, neurological deficits, and distinct dermatological features. Neurological manifestations, including seizures, intellectual disability, and various types of tumors, are often the most debilitating aspects of these syndromes, significantly impacting the quality of life of affected individuals. Early neurological signs, such as optic pathway gliomas in NF1 and cortical tubers in TSC, highlight the importance of early diagnosis and intervention. Dermatological features serve as important diagnostic clues and are often the first indication of an underlying neurocutaneous syndrome. Key skin manifestations, such as café-au-lait macules in NF1, hypomelanotic macules and facial angiofibromas in TSC, and port-wine stains in SWS, provide critical insight into the presence and severity of these conditions. The interplay between genetic mutations and their phenotypic outcomes underscores the complexity of these syndromes and the need for a multidisciplinary approach to management. Understanding the genetic and clinical spectrum of pediatric neurocutaneous syndromes is essential for improving diagnosis, treatment, and patient outcomes. Advances in molecular genetics have not only enhanced our understanding of these disorders but have also led to the development of targeted therapies, offering hope for more effective management and better quality of life for patients and their families.
Introduction
Pediatric neurocutaneous syndromes represent a group of genetically inherited disorders characterized by the simultaneous involvement of the nervous system and the skin, along with other organ systems. These syndromes are often identified by the presence of distinctive cutaneous features, which serve as important diagnostic clues. The most common neurocutaneous syndromes include Neurofibromatosis (NF), Tuberous Sclerosis Complex (TSC), and Sturge-Weber Syndrome (SWS), each associated with specific genetic mutations that lead to a spectrum of clinical manifestations. These disorders are of particular interest not only due to their complex presentations but also because they highlight the intricate relationship between genetic mutations and phenotypic outcomes. Genetic mutations in these syndromes often lead to dysregulation of cell growth and differentiation, resulting in the development of benign and malignant tumors, as well as a variety of neurological and dermatological abnormalities. For instance, Neurofibromatosis type 1 (NF1) is caused by mutations in the NF1 gene, which encodes the protein neurofibromin, a negative regulator of the Ras signaling pathway. Mutations in this gene result in the formation of neurofibromas and an increased risk of malignancies (1). Similarly, TSC is caused by mutations in the TSC1 or TSC2 genes, leading to the development of hamartomas in multiple organ systems, including the brain, skin, and kidneys (2). These genetic insights have not only advanced our understanding of the pathogenesis of these syndromes but have also opened avenues for targeted therapies.
The clinical features of neurocutaneous syndromes are highly variable and can affect multiple organ systems. Neurological manifestations, such as epilepsy, intellectual disability, and behavioral issues, are common and often the most debilitating aspects of these disorders (3). Dermatological manifestations, including café-au-lait spots, hypomelanotic macules, and angiofibromas, are among the earliest and most recognizable signs, frequently prompting further investigation into an underlying neurocutaneous syndrome. Early diagnosis is crucial as it allows for appropriate management of complications and improves the quality of life for affected individuals. This review aims to provide a comprehensive overview of pediatric neurocutaneous syndromes, with a focus on their genetic mutations, clinical features, and dermatological manifestations (4). By understanding the genetic underpinnings and phenotypic expressions of these syndromes, clinicians can improve diagnosis, management, and counseling of affected patients and their families.
Review
The genetic mutations underlying pediatric neurocutaneous syndromes are central to their diverse clinical presentations. These mutations disrupt normal cellular processes, leading to the characteristic neurological and dermatological manifestations seen in disorders such as NF, TSC, and Sturge-Weber Syndrome (SWS). The NF1 gene mutation, for example, results in the loss of neurofibromin, a key regulator of cell growth, leading to the formation of neurofibromas and an increased susceptibility to malignancies. Similarly, mutations in the TSC1 or TSC2 genes cause a loss of function in hamartin or tuberin, respectively, resulting in uncontrolled cell proliferation and the formation of hamartomas across various organ systems (5). Clinically, the neurological manifestations of these syndromes, such as seizures, developmental delays, and intellectual disabilities, often pose significant challenges in management. Dermatological features, such as hypomelanotic macules in TSC or port-wine stains in SWS, are frequently the first indicators of an underlying neurocutaneous disorder and play a crucial role in early diagnosis (6). The interplay between these genetic mutations and their phenotypic outcomes underscores the importance of a multidisciplinary approach in managing these syndromes, emphasizing the need for early detection and intervention to mitigate the impact of these lifelong conditions.
Genetic Basis of Pediatric Neurocutaneous Syndromes
The genetic basis of pediatric neurocutaneous syndromes is fundamental to understanding the diverse clinical presentations and complexities associated with these disorders. These syndromes, including NF1, TSC, and SWS, are primarily driven by specific genetic mutations that lead to the disruption of normal cellular processes, particularly those regulating cell growth and differentiation. The mutations involved in these syndromes are typically inherited in an autosomal dominant pattern, although de novo mutations are also common, contributing to the sporadic occurrence of these conditions. In Neurofibromatosis Type 1, mutations in the NF1 gene, located on chromosome 17, result in the loss of function of the neurofibromin protein. Neurofibromin is a tumor suppressor that negatively regulates the Ras signaling pathway, which is critical for controlling cell proliferation and differentiation. The absence or reduction of functional neurofibromin leads to uncontrolled cell growth, resulting in the formation of neurofibromas, café-au-lait spots, and other characteristic features of NF1. The NF1 gene is one of the most commonly mutated genes in humans, with a high mutation rate that explains the relatively high incidence of NF1 in the population (7).
Tuberous Sclerosis Complex is caused by mutations in either the TSC1 or TSC2 genes, which encode the proteins hamartin and tuberin, respectively. These proteins form a complex that inhibits the mechanistic target of rapamycin (mTOR) pathway, a key regulator of cell growth, proliferation, and metabolism. Mutations in TSC1 or TSC2 lead to hyperactivation of the mTOR pathway, resulting in the development of hamartomas in various tissues, including the brain, skin, and kidneys. The genetic heterogeneity of TSC, with mutations occurring in two different genes, contributes to the variability in clinical manifestations observed among patients (8). SWS, in contrast, is typically associated with a somatic mosaic mutation in the GNAQ gene, which encodes a G-protein subunit involved in intracellular signaling pathways. The mutation leads to abnormal blood vessel development, resulting in the characteristic port-wine stains and leptomeningeal angiomas seen in SWS. Unlike NF1 and TSC, SWS is not inherited but arises from postzygotic mutations, which result in the mosaic distribution of affected tissues (9). This somatic mutation model explains the localized nature of the vascular abnormalities and the variability in clinical severity observed in SWS. The identification of these genetic mutations has not only enhanced the understanding of the pathophysiology of pediatric neurocutaneous syndromes but has also paved the way for the development of targeted therapies. By elucidating the molecular mechanisms underlying these disorders, researchers are increasingly able to develop treatments that specifically target the affected pathways, offering hope for more effective management of these complex conditions.
Clinical Spectrum and Neurological Manifestations
The clinical spectrum of pediatric neurocutaneous syndromes is broad and complex, reflecting the diverse range of genetic mutations that underlie these disorders. Among the most significant clinical manifestations are neurological symptoms, which often present early in life and can have profound effects on the patient’s development and quality of life. These neurological manifestations are typically the result of abnormal cell growth and differentiation in the nervous system, leading to the formation of tumors, structural abnormalities, and disruptions in normal neurological function.
In NF1, one of the hallmark neurological manifestations is the development of optic pathway gliomas, which occur in approximately 15-20% of children with NF1. These low-grade tumors can lead to vision loss and, in some cases, require intervention to prevent further complications. Additionally, patients with NF1 are at an increased risk for developing other types of brain tumors, including pilocytic astrocytomas and brainstem gliomas. Beyond tumors, NF1 is also associated with learning disabilities, attention deficit hyperactivity disorder (ADHD), and seizures, all of which contribute to the significant neurodevelopmental burden seen in these patients (10). TSC presents with a distinctive neurological profile characterized by the presence of cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas (SEGAs). These structural abnormalities can lead to seizures, which are among the most common and challenging manifestations of TSC, affecting up to 90% of patients. Seizures in TSC can be refractory to treatment and may begin in infancy as infantile spasms, which are associated with poor neurodevelopmental outcomes. Intellectual disability is also common in TSC, with severity ranging from mild learning difficulties to profound cognitive impairment. Behavioral issues, including autism spectrum disorder (ASD) and aggressive behaviors, further complicate the clinical management of TSC and highlight the need for a multidisciplinary approach to care (11).
SWS primarily affects the central nervous system through the presence of leptomeningeal angiomas, which are abnormal blood vessels that can lead to chronic hypoxia and subsequent brain damage. The neurological manifestations of SWS often include seizures, which may be focal and drug-resistant, as well as hemiparesis, developmental delays, and cognitive impairments. The severity of neurological symptoms in SWS is closely related to the extent and location of the leptomeningeal angiomas, with more extensive involvement leading to more severe neurological deficits. In some cases, glaucoma may also develop due to the vascular abnormalities associated with SWS, further complicating the clinical picture (12). The neurological manifestations of pediatric neurocutaneous syndromes are diverse and often severe, necessitating early diagnosis and comprehensive management to optimize outcomes. Understanding the underlying genetic and pathophysiological mechanisms is crucial for developing effective treatment strategies and improving the quality of life for affected patients.
Dermatological Features and Their Diagnostic Significance
Dermatological manifestations are often the earliest and most recognizable signs of pediatric neurocutaneous syndromes, playing a crucial role in the initial diagnosis and ongoing monitoring of these disorders. The skin, as an accessible and visible organ, provides valuable diagnostic clues that can lead to the identification of underlying genetic conditions, making it essential for clinicians to be familiar with the characteristic cutaneous features of these syndromes.
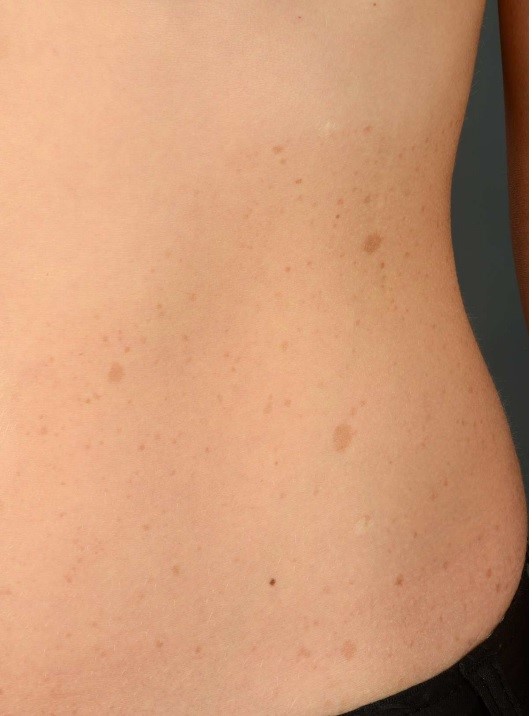
The presence of café-au-lait macules is one of the most common and earliest dermatological signs in NF1, typically appearing within the first few years of life. These hyperpigmented lesions, which are light brown and well-demarcated, are often the first clue that prompts further investigation into a potential diagnosis of NF1 (Figure 1).
Figure 1: dermatological presentation of café-au-lait macules in NF (13).
The presence of six or more café-au-lait macules greater than 5 mm in prepubertal children or 15 mm in postpubertal individuals is a major diagnostic criterion for NF1. In addition to café-au-lait macules, other dermatological features include axillary or inguinal freckling and cutaneous neurofibromas, which are benign nerve sheath tumors that can develop anywhere on the body. The number and size of neurofibromas tend to increase with age, and while they are generally benign, they can cause significant cosmetic concerns and, in some cases, discomfort or functional impairment (14).
TSC is another neurocutaneous syndrome with distinctive dermatological features that are crucial for diagnosis. Hypomelanotic macules (Figure 2), also known as ash leaf spots, are among the most common skin manifestations and can be present at birth or develop in early childhood. These hypopigmented lesions are often best visualized under Wood’s lamp examination. Facial angiofibromas, another key dermatological feature of TSC, typically appear in early childhood and are characterized by small, reddish papules distributed symmetrically across the cheeks and nose. These angiofibromas, along with other cutaneous manifestations such as shagreen patches and ungual fibromas, are highly specific for TSC and serve as important diagnostic markers. The presence of these skin lesions, particularly in combination, often leads to the consideration of TSC in the differential diagnosis of children presenting with seizures or developmental delays (8).
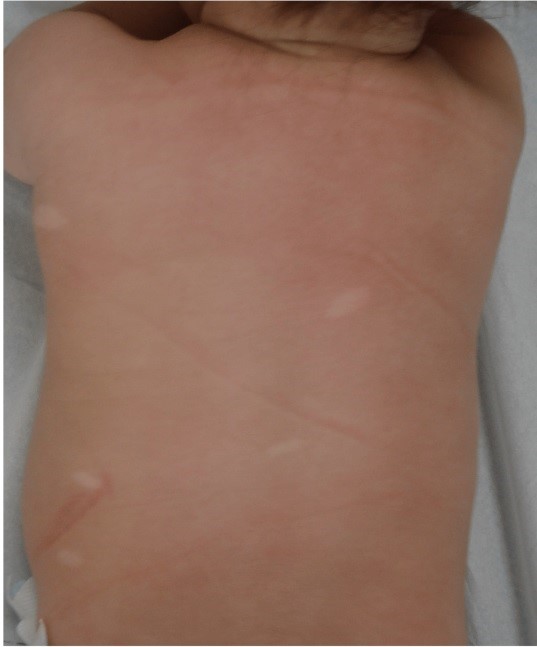
Figure 2: Ash leaf spot hypomelanotic lesion in a 7 year-old boy’s arm (15).
SWS is distinguished by the presence of a port-wine stain, a congenital capillary malformation that affects the skin and is often the first sign of the syndrome. This vascular anomaly is typically unilateral and follows the distribution of the trigeminal nerve. The port-wine stain may darken and thicken over time, and its presence, especially when located on the face, should prompt further investigation for potential neurological involvement, such as leptomeningeal angiomas. The recognition of this distinctive dermatological feature is critical for the early diagnosis and management of SWS, as timely intervention can help mitigate the associated neurological complications (16). In summary, the dermatological features of pediatric neurocutaneous syndromes not only aid in the diagnosis of these complex disorders but also provide insight into the severity and progression of the disease. Early recognition and thorough skin examination are essential components of the clinical evaluation, guiding further diagnostic testing and management strategies.
Conclusion
Pediatric neurocutaneous syndromes are complex disorders rooted in specific genetic mutations that lead to diverse neurological and dermatological manifestations. Early recognition of these features is critical for diagnosis and effective management. Advances in genetic understanding have paved the way for targeted therapies, offering improved outcomes for affected patients. A multidisciplinary approach remains essential to optimizing care and enhancing quality of life.
Disclosures
Author Contributions
The author has reviewed the final version to be published and agreed to be accountable for all aspects of the work.
Ethics Statement
Not applicable
Consent for publications
Not applicable
Data Availability
All data is provided within the manuscript.
Conflict of interest
The authors declare no competing interest.
Funding
The author has declared that no financial support was received from any organization for the submitted work.